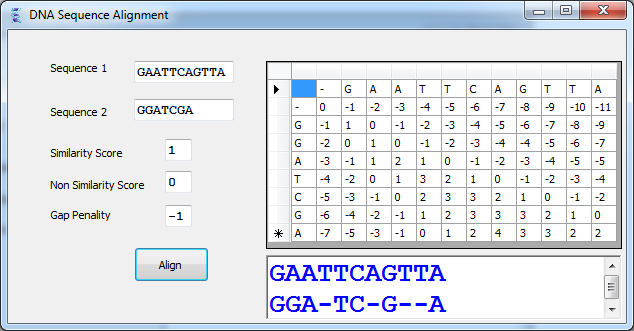
Introduction
Sequence alignment is the procedure of comparing two (pair-wise alignment) or more (multiple alignment) sequences by searching for a series of characters that are in the same order in all sequences. Two sequences can be aligned by writing them across a page in two rows. Identical or similar characters are placed in the same column, and non identical ones can either be placed in the same column as a mismatch or against a gap (-) in the other sequence. Sequences that are aligned in this manner are said to be similar. Sequence alignment is useful for discovering functional, structural, and evolutionary information in biological sequences. Consider the following DNA Sequences GACGGATTAG and GATCGGAATAG. Notice that when we align them one above the other:
GA-CGGATTAG
GATCGGAATAG
The only differences are marked with colors in the above sequences. Observe that the gap (-) is introduced in the first sequence to let equal bases align perfectly. the goal of this article is to present an efficient algorithm that takes two sequences and determine the best alignment between them. The total score of the alignment depends on each column of the alignment. If the column has two identical characters, it will receive value +1 (a match). Different characters will give the column value -1 (a mismatch). Finally a gap in a column drops down its value to -2 (Gap Penalty). The best alignment will be one with the maximum total score. The above alignment will give a total score: 9 × 1 + 1 × (-1) + 1 × (-2) = 6.
These parameters match, mismatch and gap penalty can be adjusted to different values according to the choice of sequences or experimental results.
One approach to compute similarity between two sequences is to generate all possible alignments and pick the best one. However, the number of alignments between two sequences is exponential and this will result in a slow algorithm so, Dynamic Programming is used as a technique to produce faster alignment algorithm. Dynamic Programming tries to solve an instance of the problem by using already computed solutions for smaller instances of the same problem. Giving two sequences Seq1 and Seq2 instead of determining the similarity between sequences as a whole, dynamic programming tries to build up the solution by determining all similarities between arbitrary prefixes of the two sequences. The algorithm starts with shorter prefixes and uses previously computed results to solve the problem for larger prefixes.
Let M =size of Seq1 and N= size of Seq2 ,the computation is arranged into an (N+1) × (M+1) array where entry (j,i) contains similarity between Seq2[1.....j] and Seq1[1.....i]. The algorithm computes the value for entry(j,i) by looking at just three previous entries:
(j-1,i-1) Diagonal Cell to entry (j,i)
(j-1,i) Above Cell to entry (j,i)
(j,i-1) Left Cell to entry (j,i)
j-1,i
j,i
j-1,i-1
j,i-1

The value of the entry (j,i) can be computed by the following equation:
| Equation (1.1) |
where p(j,i)= +1 if Seq2[j]=Seq1[i] (match Score) and p(j,i)= -1 if Seq2[j]!=Seq1[i].
The maximum value of the score of the alignment located in the cell (N-1,M-1) and the algorithm will trace back from this cell to the first entry cell (1,1) to produce the resulting alignment . IF the value of the cell (j,i) has been computed using the value of the diagonal cell, the alignment will contain the Seq2[j] and Seq1[i]. IF the value has been computed using the above cell, the alignment will contain Seq2[j] and a Gap ('-') in Seq1[i]. IF the value has been computed using the left cell, the alignment will contain Seq1[i] and a Gap ('-') in Seq2[j]. the resulting alignment will produce completely by traversing the cell (N-1,M-1) back towards the initial entry of the cell (1,1).
Using the Code
My code has two classes, the first one named DynamicProgramming.cs and the second named Cell.cs. I will discuss the details of DynamicProgramming.cs class in the following lines because it describes the main idea of my article.
The first class contains three methods that describe the steps of dynamic programming algorithm. The first method is named Intialization_Step, this method prepares the matrix a[i,j] that holds the similarity between arbitrary prefixes of the two sequences. The algorithm starts with shorter prefixes and uses previously computed results to solve the problem for larger prefixes.
public static Cell[,] Intialization_Step
(string Seq1, string Seq2,int Sim,int NonSimilar,int Gap)
{
int M = Seq1.Length;
int N = Seq2.Length;
Cell[,] Matrix = new Cell[N, M];
for (int i = 0; i < Matrix.GetLength(1); i++)
{
Matrix[0, i] = new Cell(0, i, i*Gap);
}
for (int i = 0; i < Matrix.GetLength(0); i++)
{
Matrix[i, 0] = new Cell(i, 0, i*Gap);
}
for (int j = 1; j < Matrix.GetLength(0); j++)
{
for (int i = 1; i < Matrix.GetLength(1); i++)
{
Matrix[j, i] = Get_Max(i, j, Seq1, Seq2, Matrix,Sim,NonSimilar,Gap);
}
}
return Matrix;
}
The second method named Get_Max computes the value of the cell (j,i) by the Equation 1.1 .
public static Cell Get_Max(int i, int j, string Seq1,
string Seq2, Cell[,] Matrix,int Similar,int NonSimilar,int GapPenality)
{
Cell Temp = new Cell();
int Sim;
int Gap = GapPenality;
if (Seq1[i] == Seq2[j])
Sim = Similar;
else
Sim = NonSimilar;
int M1, M2, M3;
M1 = Matrix[j - 1, i - 1].CellScore + Sim;
M2 = Matrix[j, i - 1].CellScore + Gap;
M3 = Matrix[j - 1, i].CellScore + Gap;
int max = M1 >= M2 ? M1 : M2;
int Mmax = M3 >= max ? M3 : max;
if (Mmax == M1)
{ Temp = new Cell(j, i, M1, Matrix[j - 1, i - 1],
Cell.PrevcellType.Diagonal); }
else
{
if (Mmax == M2)
{ Temp = new Cell(j, i, M2, Matrix[j, i - 1], Cell.PrevcellType.Left); }
else
{
if (Mmax == M3)
{ Temp = new Cell(j, i, M3, Matrix[j - 1, i],
Cell.PrevcellType.Above); }
}
}
return Temp;
}
The third method is named Traceback_Step. This method will produce the alignment by traversing the cell matrix(N-1,M-1) back towards the initial entry of the cell matrix (1,1).
public static void Traceback_Step(Cell[,] Matrix,
string Sq1, string Sq2, List<char> Seq1, List<char> Seq2)
{
Cell CurrentCell = Matrix[Sq2.Length - 1, Sq1.Length - 1];
while (CurrentCell.CellPointer != null)
{
if (CurrentCell.Type == Cell.PrevcellType.Diagonal)
{
Seq1.Add(Sq1[CurrentCell.CellColumn]);
Seq2.Add(Sq2[CurrentCell.CellRow]);
}
if (CurrentCell.Type == Cell.PrevcellType.Left)
{
Seq1.Add(Sq1[CurrentCell.CellColumn]);
Seq2.Add('-');
}
if (CurrentCell.Type == Cell.PrevcellType.Above)
{
Seq1.Add('-');
Seq2.Add(Sq2[CurrentCell.CellRow]);
}
CurrentCell = CurrentCell.CellPointer;
}
}
The second class in my code is named Cell.cs. This class manipulates the cell of the matrix. Each cell has:
- A location indicated by the index of the row and index of the column
- A value that is represented by the score of the alignment
- A pointer to a previous cell that is used to compute the score of the current cell [Note: Pointer value "Diagonal, Above and Left"]
Reference
- "Introduction To Computational Molecular Biology" by JOÃO SETUBAL and JOÃO MEIDANIS

