This is about a Huffman-Encoded bit stream cached inside system's combined video-memory, indexed and mapped through pcie bridges, for saving disk drive bandwidth for other tasks and saving available RAM for critical tasks.
Introduction
When a big nucletide sequence file (such as in FASTA format) is needed to build a list of base-pair sequences for random-access for an application, it may need more space than the available RAM of system. Sometimes, there is no empty RAM slot to upgrade to test a prototype nucleotide sequence matching algorithm on a big data set. But there are libraries to index relevant data from disk drives directly without consuming the available RAM. This tool, on the other hand, does a similar thing on graphics card memory with help of Huffman Encoding Algorithm and OpenCL data transferring. It maps well to asymmetrical systems with low RAM + high VRAM or cheap HDD + cheap GPU + expensive CPU.
Background
The main parts of the sequence caching algorithm are:
- full data compression for both sequences and descriptors
- data distribution to multiple cards for increasing bandwidth & capacity
- partial decompression when accessing an index of sequence or descriptor
Compression algorithm is the Huffman Encoding. First, all the data is parsed for counting number of occurrences for each symbol. For bioinformatics, it's at least consisted of 'A','C','G' and 'T'. Some files have more symbols but having just 4 letters makes it ~1/4 size after compression.
After the counting is done (which is the pass 1), a Huffman Tree is built. It is done like this:
- Sort all occurrence counts in increasing order
- Take first 2 occurrences and put them in a new occurrence "node" (which has some other members for book keeping when encoding & decoding too)
- Put this new node back in same array (also works with a priority queue instead of array)
- Sort array again
- Repeat until there is only 1 node left in array
Actual codes that build the Huffman Tree on a std::vector:
while(sortedNodes.size()>1)
{
Node<T> node1 = sortedNodes[0];
Node<T> node2 = sortedNodes[1];
Node<T> newNode;
newNode.count = node1.count + node2.count;
newNode.data=0;
newNode.leaf1 = node1.self;
newNode.leaf2 = node2.self;
newNode.self = ctr;
newNode.isLeaf=false;
sortedNodes.erase(sortedNodes.begin());
sortedNodes.erase(sortedNodes.begin());
sortedNodes.push_back(newNode);
referenceVec.push_back(newNode);
std::sort(sortedNodes.begin(), sortedNodes.end(),
[](const Node<T> & n1, const Node<T> & n2){ return n1.count<n2.count;});
ctr++;
}
root = sortedNodes[0];
This result node element is called root and is used as entrance point for a path of decoding for a symbol.
template<typename T>
struct Node
{
size_t count;
int self;
int leaf1;
int leaf2;
T data;
bool isLeaf;
};
This makes it easy to find a symbol from its binary encoded data path. Root node has no bit. But leaf1 is named as '0' and leaf2 is named as '1'. So, when reading a compressed data, if first element is '1', then the leaf2 node index is selected. After that, next bit is read from compressed data. Then, again, leaf1 or leaf2 path is taken, until isLeaf is true. When isLeaf is true, the selected node is the last node in path and contains the decoded symbol (like 'A', 'C', ..) in data member.
"Path" of each symbol is computed by a recursive function that holds temporary std::vector<bool> variable for each depth of tree traversal. Every new depth adds a new bit to the vector and the result vector is assigned to another data structure for access during decoding.
Normally, just count, leaf1 and leaf2 are enough to work for both encoding and decoding. But to optimize for speed, isLeaf member is used as a shortcut instead of checking both leaf1 and leaf2 in decoding. Data type is unsigned char which means byte-level compression is done here. To increase compression ratio, maybe in future, a pack of bytes, like a short or an int or even a size_t can be used instead. But increasing compression resolution also increases tree size for less-compression-friendly data. For example, if a FASTA file has more than just "ACGT" letters, then number of leaf nodes in Huffman Tree can grow up to 256 * 256 * 256 * 256 nodes for int-based compression. This is not good for cached access and defeats the purpose of compression (saving bandwidth & memory capacity through computational effort). For now, only unsigned char is used and the Huffman Tree contains at most several kB of Node data.
In the next pass (pass 2) for the file, its total bitcount is deduced. Since RAM may not be available to hold big file, an extra pass is required to count bits of compressed data. Both descriptors and sequences of FASTA file are compressed so there is a big bit stream holding both sequence bitstream and descriptor bitstream. But both have their own unique Huffman Tree so that while descriptors usually have 256 characters with less compression, sequence part can still be compressed as good as its own symbol table (Just A and B letters compressed to 1 and 0, 8x compression). To count bits, each letter is mapped to a bit stream size. For example, if Adenine nucleobase ('A' symbol) has Huffman Tree path of '1 - 1 - 0', then its bit stream has 3 elements. Number of bits is increased by 3 for 'A'.
Once bitcount is known, a video-memory backed virtual array is allocated (that has static size and interleaved access to graphics card as some kind of "paging" that does thread-safe access to any data and leverage high bandwidth advantage of pcie in multithreaded access). (How it works: VirtualMultiArray - A C++ Virtual Array Implementation)
Pass 3 starts if virtual array is successfully allocated. This time, instead of counting bits, those bits are copied to the virtual array. Every bit is put inside an unsigned char element's relevant bit position. First bit goes into first bit of first byte, second bit goes into second bit of first byte, ... it is completed when the last bit goes to last byte's k-th bit position.
Reading a big file 3 times is slow. This is only for initialization. Once it is complete, accessing a sequence takes microseconds of pcie data copy and microseconds of Huffman Decoding (and increases linearly with size of required sequence).
Each access to a sequence index goes through VRAM-cached "backing store", RAM-cached "page" and decoding (in L1 cache for small sequence, RAM for big sequence). For single thread access, these are sequential tasks. In multithreaded access, these latencies are hidden behind each other so that while one thread loads data from VRAM, another thread loads data from RAM and another thread computes Huffman Decoding.
Using the Code
To start a minimal example that reads a sequence from a FASTA formatted file, at least a few things need to be prepared after getting header from repository (https://github.com/tugrul512bit/FastaGeneIndexer).
First, internal virtual array implementation (VirtualMultiArray, https://github.com/tugrul512bit/VirtualMultiArray) uses OpenCL. This needs the linker to link to its driver library (/usr/local/cuda/lib64 folder for Ubuntu+Nvidia, system32 folder Windows) and include the relevant include directory for using OpenCL data copies.
Second, it uses OpenMP in one of parsing phases. This needs relevant OpenMP flags enabled for compiler and linker. Some compilers have this built in already.
Lastly, it uses C++17 to get file size and C++14 for other parts and project needs to be 64-bit (its default in g++10 unless stated otherwise, also works with g++7.5 and msvc 2019).
In development computer (Ubuntu 18.04, Nvidia Nsight, g++10), these are the compiling & linking commands:
g++-10 -std=c++17 -O3 -g3 -Wall -c -fmessage-length=0 -mavx -march=native
-funroll-loops -mtune=native -funsafe-math-optimizations -ftree-vectorize
-ffast-math -fopenmp -pthread -fPIC -MMD -MP -MF"src/main.d" -MT"src/main.d"
-o "src/main.o" "../src/main.cpp"
g++-10 -L/usr/local/cuda/lib64 -o "Virtualizer2" ./src/main.o -lOpenCL -lgomp -lpthread
Other than the VirtualMultiArray headers needed, it has only single header for everything else:
#include"FastaGeneIndexer.h"
Reading a FASTA file takes no more than one liner:
FastaGeneIndexer cache("./data/influenza.fna");
If file parsing passes & Huffman Tree contents need to be seen, a debug flag is given:
bool debug = true;
FastaGeneIndexer cache("./data/influenza.fna", debug);
In case of invalid parameters for virtual array / opencl api, it throws exceptions, so that they can be caught like this:
try
{
bool debug = true;
FastaGeneIndexer cache("./data/influenza.fna", debug);
}
catch(std::exception & e)
{
std::cout<< e.what() <<std::endl;
}
Then sequences can be read:
#include "FastaGeneIndexer.h"
int main(int argC, char** argV)
{
try
{
bool debug = true;
FastaGeneIndexer cache("./data/influenza.fna", debug);
std::string sequence = cache.getSequence(5);
std::string descriptor = cache.getDescriptor(5);
std::cout<< sequence <<std::endl;
std::cout<< descriptor <<std::endl;
}
catch(std::exception & e)
{
std::cout<< e.what() <<std::endl;
}
return 0;
}
Output
ATGAGTCTTCTAACCGAGGTCGAAACGTACGTTCTCTCTATCGTCCCGTCAGGCCCCCTCAAAGCCGAGATCGCACAGAGACTT
GAAGATGTCTTTGCAGGGAAGAACACCGATCTTGAGGCTCTCATGGAATGGCTAAAGACAAGACCAATCCTGTCACCTCTGACT
AAGGGGATTTTAGGATTTGTGTTCACGCTCACCGTGCCCAGTGAGCGGGGACTGCAGCGTAGACGCTTTGTCCAAAATGCTCTT
AATGGGAACGGAGATCCAAATAACATGGACAAAGCAGTTAAACTGTATAGGAAGCTTAAGAGGGAGATAACATTCCATGGGGCC
AAAGAAATAGCACTCAGTTATTCTGCTGGTGCACTTGCCAGTTGTATGGGCCTCATATACAACAGGATGGGGGCTGTGACCACT
GAAGTGGCATTTGGCCTGGTATGCGCAACCTGTGAACAGATTGCTGACTCCCAGCATCGGTCTCATAGGCAAATGGTGACAACA
ACCAATCCACTAATCAGACATGAGAACAGAATGGTTCTAGCCAGCACTACAGCTAAGGCTATGGAGCAAATGGCTGGATCGAGT
GAGCAAGCAGCAGAGGCCATGGATATTGCTAGTCAGGCCAGGCAAATGGTGCAGGCGATGAGAACCATTGGGACTCATCCTAGC
TCCAGTGCTGGTCTAAAAGATGATCTTCTTGAAAATTTGCAGGCCTATCAGAAACGAATGGGGGTGCAGATGCAACGATTCAAG
TGATCCTCTCGTTATTGCAGCAAATATCATTGGGATCTTGCACTTGATATTGTGGATTCTTGATCGTCTTTTTTTCAAATGCAT
TTATCGTCGCTTTAAATACGGTTTGAAAAGAGGGCCTTCTACGGAAGGAGTGCCAGAGTCTATGAGGGAAGAATATCGAAAGGA
ACAGCAGAATGCTGTGGATGTTGACGATGGTCATTTTGTCAACATAGAGCTGGAGTAA
gi|60460|gb|X08089|Influenza A virus (A/WS/7/1933(H1N1))
ORF1 for M1 protein and ORF2 for M2 protein, genomic RNA
FastaGeneIndexer constructor with a file name string parameter initializes everything needed for accessing descriptors and sequences. Parsing file, building Huffman Tree, allocating virtual array, everything is complete in constructor. Doing multiple passes require some time for big input files so enabling debugging helps to see if there is any progress in preparing compression is made.
There is no need to explicitly deallocate anything. Destructors of internal data structures automatically release all resources like OpenCL buffers, pages and Huffman nodes.
Maximum bandwidth relies heavily on multi-threaded access and presence of multiple graphics cards and single-thread performance of processor.
With simple benchmarking for first access on single-thread:
#include "FastaGeneIndexer.h"
#include "CpuBenchmarker.h"
int main(int argC, char** argV)
{
try
{
bool debug = true;
FastaGeneIndexer cache("./data/influenza.fna", debug);
{
CpuBenchmarker bench(0,"read 1");
cache.getSequence(10);
}
}
catch(std::exception & e)
{
std::cout<< e.what() <<std::endl;
}
return 0;
}
Output
read 1: 104912 nanoseconds
This is mostly due to using OpenCL data copying done first time (and causing drivers to lazy-initialize some internal state in somewhere) and getting data from video memory into a page (which is a RAM-cache). Next reading has better performance:
{
CpuBenchmarker bench(0,"read 1");
cache.getSequence(10);
}
{
CpuBenchmarker bench(0,"read 2 same location");
cache.getSequence(10);
}
Output
read 1: 148684 nanoseconds
read 2 same location: 77568 nanoseconds
Just 2 access may not be showing real world performance. CPU also needs to be in higher frequency states for a better benchmarking:
for(int i=0;i<10000;i++)
{
cache.getSequence(i);
}
{
CpuBenchmarker bench(0,"read 10k sequences, single thread",10000);
for(int i=0;i<10000;i++)
{
cache.getSequence(i);
}
}
Output
read 1: 145723 nanoseconds
read 2 same location: 78204 nanoseconds
read 10k sequences, single thread: 300136627 nanoseconds
(throughput = 30013.66 nanoseconds per iteration)
This is on FX8150@2.1GHz + 1 channel DDR3 1333MHz RAM + (pcie v2.0 x8 + x4 + x4). The better the core performance of cpu & RAM latency, the lower the decoding latency. Decoding means getting data from graphics card and some tree traversal on Huffman Tree and moving data between containers multiple times.
The main idea is to do concurrent accesses to the cache to have more bandwidth than an old HDD:
for(int i=0;i<10000;i++)
{
cache.getSequence(i);
}
size_t count=0;
{
CpuBenchmarker bench(0,"read 10k sequences, single thread",10000);
for(int i=0;i<10000;i++)
{
count += cache.getSequence(i).size();
}
}
{
CpuBenchmarker bench(count,"read 10k sequences, multi-thread",10000);
#pragma omp parallel for
for(int i=0;i<10000;i++)
{
cache.getSequence(i);
}
}
Output
read 1: 146718 nanoseconds
read 2 same location: 78615 nanoseconds
read 10k sequences, single thread: 302150806 nanoseconds
(throughput = 30215.08 nanoseconds per iteration)
read 10k sequences, multi-thread: 55878726 nanoseconds
(bandwidth = 203.29 MB/s) (throughput = 5587.87 nanoseconds per iteration)
Scaling on average time per read is less than number of cores (8) but greater than number of core-modules (4) and single channel memory at 1333MHz has some effect in this. To see it better, setting CPU frequency back to default(3.6GHz) (after 47 seconds of initialization):
read 1: 154930 nanoseconds
read 2 same location: 79527 nanoseconds
read 10k sequences, single thread: 177180777 nanoseconds
(throughput = 17718.08 nanoseconds per iteration)
read 10k sequences, multi-thread: 38953637 nanoseconds
(bandwidth = 291.62 MB/s) (throughput = 3895.36 nanoseconds per iteration)
Average access time improved by 43% with 71% higher cpu frequency. Single-thread access latency improved by 70%. This is much closer to a memory bandwidth bottleneck caused by single channel DDR3 at 1333MHz. RAM bandwidth is shared between PCI-E data transfers and decoding. Too long decoding sequence means higher data moved between containers in getSequence() and less bandwidth left for PCI-E transfers. For this reason, some FASTA files with just 5-10 sequences (but each being megabytes-long) leave not much multi-threading advantage (for current version) but still have better performance than single thread.
Different systems would have different bottlenecks. A Threadripper may be bottlenecked on memory too while an Epyc may have pcie bottleneck with 1-2 cards.
Sample file was 1.4GB and only 15% of system's video memory was used during tests.
Features
Sub-Sequence Query
User can also query for K number of nucleotides of a sequence.
This overloaded method (of FastaGeneIndexer::getSequence(index)) takes two extra parameters. Starting position of sub-sequence within the selected sequence and length of it.
FastaGeneIndexer cache("./data/yersinia_pestis_genomic.fna");
std::cout << cache.getSequence(3,100,100) << std::endl;
Output
GACAGTTATGGAAATTAAAATCCTGCACAAGCAGGGAATGAGTAGCCGGGCGATTGCCAGAGAACTGGGGAT
CTCCCGCAATACCGTTAAACGTTATTTG
It does not write it with line-feed characters. All the data stored in graphics cards is pure nucleotide names and descriptor strings.
Since this method does not fetch full sequence data, it completes much quicker. Single symbol query performance for FX8150 at 3.6GHz:
sub-sequence query (sequential-access latency benchmark, single thread):
5603564809 nanoseconds
(throughput = 1160.19 nanoseconds per iteration)
sub-sequence query (random-access latency benchmark, single thread):
5432723756 nanoseconds
(throughput = 1167.39 nanoseconds per iteration)
1000-symbol query performance with multi-threading:
bandwidth benchmark, sequential access, multi-threaded:
166062 nanoseconds
(bandwidth = 423.37 MB/s) (throughput = 2372.31 nanoseconds per iteration)
(just 150 microseconds of benchmarking contains some noise)
bandwidth benchmark, random access, multi-threaded:
157730 nanoseconds
(bandwidth = 445.73 MB/s) (throughput = 2253.29 nanoseconds per iteration)
Sequence & Sub-Sequence Query by a Descriptor Name
After calling
FastaGeneIndexer::initDescriptorIndexMapping()
method once, user can fetch a (sub/full)sequence by its descriptor name.
This method takes a string parameter as descriptor name and rest of the parameters are the same as sub-sequence query.
<code>FastaGeneIndexer::getSequenceByDescriptor(std::string, size_t, size_t)</code>
bool debug = true;
FastaGeneIndexer cache(<span class="pl-pds">"./data/influenza.fna"</span>, debug);
cache.initDescriptorIndexMapping();
std::cout << cache.getSequenceByDescriptor(cache.getDescriptor(1),50,50) << std::endl;
Output
CGTACGTTCTCTCTATCGTCCCGTCAGGCCCCCTCAAAGCCGAGATCGCA
Total Symbol Count for Whole File
User can get precomputed values of symbol counts.
FastaGeneIndexer::getSymbolCount(unsigned char)
bool debug = true;
FastaGeneIndexer cache("./data/influenza.fna", debug);
std::cout << cache.getSymbolCount('A') << std::endl;
std::cout << cache.getSymbolCount('C') << std::endl;
std::cout << cache.getSymbolCount('G') << std::endl;
std::cout << cache.getSymbolCount('T') << std::endl;
Output
437637159
249039370
309033962
304296666
Total Number of Indexed Sequences
The method FastaGeneIndexer::n() returns number of indexed (and cached) sequences. For example, influenza.fna has more than 800k sequences in 1.4GB file.
Parallel Sequence Query
FastaGeneIndexer::getSequenceParallel(size_t index) is optimized for fetching very long sequences (as in homo sapiens chromosome's 250MB+ per sequence) quickly. It uses OpenMP under the hood for a very simple for loop that fetches chunks of data in parallel and concatenates them as a result a string. Since decoding takes time, it uses all of cores for a good speedup. Doing the work in smaller chunks (which is pageSize sized or 256k for big files) makes use of cache and increases overall read performance.
Optional Extra RAM Usage
With third constructor parameter for FastaGeneIndexer class set to true, caching with more RAM is enabled to let many-core CPUs be able to overlap sequence queries, to increase overall throughput.
Optionally Limit First Graphics Card's VRAM Usage
On multi-gpu systems, first/main card is always used by OS and loses ~400 MB (on 2GB cards) of its VRAM to serve for OS functions. Due to this, a fourth parameter was added to FastaGeneIndexer constructor. When this parameter is given true, caching allocates 25% less VRAM space on main card and lets other cards fully fill their VRAM with Huffman Encoded data. For example, when caching 4 x 3.2GB chromosome.fa files on a 3 x 2GB card system, enabling this parameter fills 85% of first card instead of overflowing. Two other cards use 95% of their VRAM without problems.
How To Find Common Sequences Between Several FASTA Files?
It's just like finding intersection between two vectors of integers with extra book-keeping for lengths of each sequence. Here, we are finding exact same nucleobase sequences (each sequence is with its own descriptor but this example did not take descriptor data into consideration. Just sequence part.) between 4 files each with 3.2GB data on 194 sequences (194 x 4 sequences total, 12.8GB total).
Algorithm:
- Sort all sequence indices on their lengths, for each file independently.
- Pick first two sorted index arrays.
- Do the "intersection algorithm"
- while ( both arrays counters in range )
- {
- find a point where both sequences have equal length
- scan whole block of same length, for both arrays
- brute force comparison (for all elements of two blocks)
- fetch x from array 1 block
- fetch y from array 2 block
- compare
- if equal, write to result map (
std::map)
- }
- Repeat N-1 times but this time using "result (
std::map)" that contains intersection elements and next file.
- use result2 map as output too, to keep old values of "result"
- do ping-pong between result and result2 to keep intermediate results
- When all files are compared, look for result or result2 array (depending on number of files being even or odd)
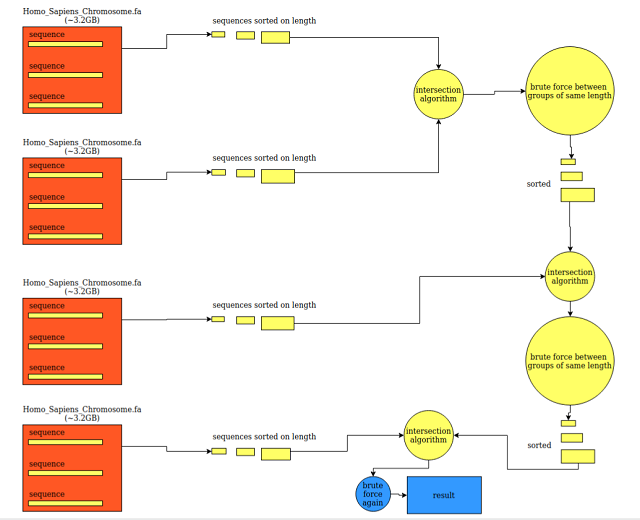
You can find test function codes here: https://github.com/tugrul512bit/FastaGeneIndexer/blob/main/test_find_common_sequences.cpp
As you can see from the code in the link, the sorting is not actually in-place. This tool is only a read-only cache. The sorting is made out-of-array with just sorting indices, without moving a single data byte. Just movement of indices on a second layer of indirection (std::vector, std::map). Here, std::map is very useful to hash collisions between arrays because it removes duplicates automatically without any checks.
For the development computer which has only 4GB RAM, 6GB (combined) VRAM and FX8150(at 3.6GHz), the benchmarking results are:
- Initialization time = 181 seconds = 70 MB/s average HDD-read + Huffman Encoding throughput
- Parsing only 4 files lets only 4 threads to be used
- Partially slowed by old HDD having maximum ~150 MB/s read throughput (only for sequential read)
- Partially speed up by Ubuntu's own file cache
- Mainly encoding bottleneck
- Compute time = 72 seconds = 355 MB/s average VRAM data fetch + Huffman Decoding + comparing
- Uses all threads of CPU
- Mainly bottlenecked by decoding
- The more cards the better
- Comparison of strings is just
== which is optimized by compiler and complete quickly - Assuming linear scan for 355 MB/s value. If there is too much brute-force happening, then actual bandwidth is higher
- System RAM is single channel 1333MHz = 10GB/s = this is absolute maximum RAM-based scanning bandwidth with actually doing nothing useful. Once
std::string data is copied between containers, bandwidth is lost. Const-correctness in query method helps in optimizing this part.
- cpu: hot (85%+ usage in compute)
- system: totally responsive
Points of Interest
Keeping Huffman Tree as a combination of Tree of nodes & pointers was slower than using a vector of nodes and integer indices. This was probably due to size of pointers (8 bytes in system) being twice that of just integer indexing amd jumping to far uncached locations. Even a 16 bit index can be used since this is only a char-level compression with no more than 64k nodes needed at any time. But 32bit integer (or whatever suitable size for system) is good enough when following bit after bit in the compressed stream. Leaf1 - leaf2 indices point to elements of vector of nodes. It helps cached access to nodes without jumping far locations in memory.
After speed-related optimizations, std::map (to count symbol occurences) became the bottleneck in first 2 file parsing passes. Instead of mapping like:
std::map<unsigned char, size_t> counts;
if(counts.find(symbol)==counts.end())
{
counts[symbol]=1;
}
else
{
counts[symbol]++;
}
using simple array is much faster:
unsigned char counts[256];
counts[symbol]++
Similarly, keeping compressed bits (std::vector<bool>) of each symbol in std::map is much slower than using simple array of 256 size. Increasing memory consumption a bit gave much better parsing performance.
Learning Huffman Encoding is a nice experience and for text compression, even on byte-basis, it is very good. For just "ACTG" combinations, it makes up to ~3.9x compression. For full alphabet and a bit of other symbols included, it still compresses from ~600MB to ~450MB which is much better than nothing.
If there were words (not letters) in "sequence" part of FASTA formatted file, then it would have much better compression with a tuning of algorithm for words. Something like { 0: "hello", 1:"world" } could happen for a 40x compression. But only descriptor parts have words and descriptors are much smaller than sequences so they were not optimized for word-base Huffman Encoding and simply joined to same bit stream of sequences (but with their own Huffman Trees).
In the current version, main bottlenecks are:
- Initialization (3x file parsing, last one being longer due to Huffman Encoding)
- CPU single-thread performance (to decode using Huffman Tree)
- RAM bandwidth (which is shared between VRAM data transfers and decoding)
- PCI-E bandwidth (only when other parts are fully optimized or there are enough hardware specs for them)
Due to these, it's only useful for doing many multithreaded access to sequences for a long time. Maybe in future, it can be optimized for better compression schemes, like finding run-lengths of most probable nucleotide repetitions (like AAACCCCAAAGTAAACCCCAAAGT) and treating them as a single symbol. Once compression ratio is improved, the pcie or ram bandwidth becomes less important and cpu core performance is used better (considering decoded data fits in cache). Duplicates could be treated as a symbol, too. But finding duplicates would add another layer in initialization.
History
- 24th February, 2021: Base information added
- 25th February, 2021: Improved article presentation to include algorithms used
- 27th February, 2021: Added new features. Sub-sequence query, get total nucleotide count per symbol(A,C,G,T,...), get sequence by descriptor string
- 2nd March, 2021: Added sample algorithm for "finding common sequences of several FASTA files" and new features

