This post covers a few simple topics in Computational physics handled in Python.
Introduction
I have another repository on GitHub. I decided to have one where I’ll put Python code for computational physics issues that are simpler / less complete than the code for the C++ projects. I’ll put there both jupyter notebooks and python scripts. At this moment, there are only two of them, hopefully I’ll add more.
So, without much ado, here it is: PythonCompphys.
At this moment, only two subjects are in there, Hartree-Fock and the Car-Parrinello one.
The Hartree-Fock is a preliminary for the Car-Parrinello and a gentler approach to Hartree-Fock than the Hartree-Fock C++ project. It does not take into account symmetries and optimizations for solving the integrals do not exist and it’s only considering spherically symmetric Gaussian Orbitals.
Program in Action
This time, there is no video. You have the option of running the notebooks in binder (there is a button at the bottom of the README if you visit the GitHub repository) or better, download the notebooks or the python scripts and run them locally.
Here is a screenshot of the script run in the Qt Console:
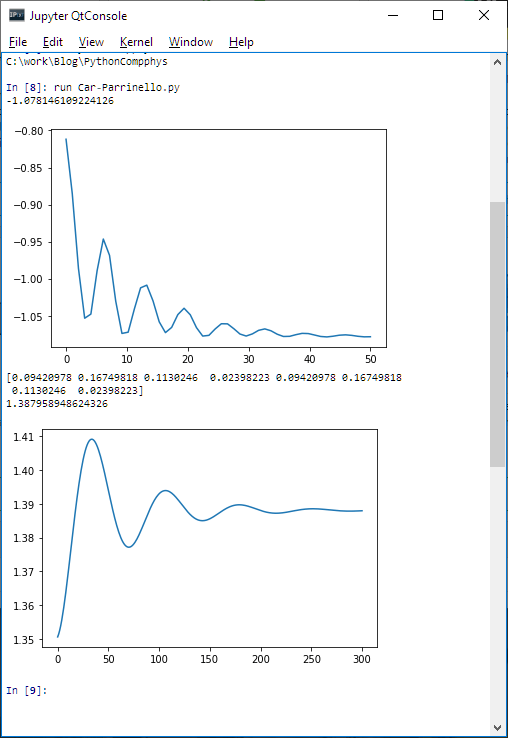
Car-Parrinello script run
In the first chart, the charted value is the energy of the H2 molecule with the distance of the nuclei being kept constant (it’s not the equilibrium one), while electrons relax towards the ground state. The result is very close to the Hartree-Fock solution.
In the second chart, both the electrons and the nuclei relax, the chart being for the distance between nuclei. The result is again quite good for the equilibrium distance, considering the used basis. Also, the frequency of oscillation is close to the real one.
Theory
The theory is very nicely exposed in Computational Physics book by Jos Thijssen, Chapter 9, Quantum Molecular Dynamics.
It’s also described in Electronic Structure, Basic Theory and Practical Methods by Richard M Martin, Chapter 18, having the same title, Quantum Molecular Dynamics.
Some Wikipedia links for start: Car–Parrinello molecular dynamics, Hellmann–Feynman theorem, Verlet integration, Euler–Lagrange equation.
And here is a lecture by Roberto Car (Princeton University) and Michele Parrinello (ETHZ) at CECAM:
Here are some slides for a lecture describing Car-Parrinello: Ab initio molecular dynamics from University of Southampton. I’m sure there are many others that are available, I won’t provide a lot of more links here.
One more is of course worth mentioning, the original article of Car and Parrinello: Unified Approach for Molecular Dynamics and Density-Functional Theory Phys. Rev. Lett. 55, 2471 – Published 25 November 1985.
The main ideas of the method are:
- separate out the electrons and nuclei dynamics, Born–Oppenheimer style
- use the Euler–Lagrange equations, but for the electrons, instead of considering the electrons coordinates as dynamic variables, as it’s usual in the classical cases, consider instead the expansion coefficients of the state in the used basis as the dynamic variables
- add the constraint of orthonormality using Lagrange multipliers
- use Hellman-Feynman theorem to compute the forces
- use a fictional ‘friction’ force that allows the system to lose energy and evolve towards the ground state
- in this case, Verlet integration is used to integrate the equations of motion
The Code
The code is relatively simple and it uses only math, numpy, scipy and matplotlib for charts.
This is probably the most important part:
for cycle in range(numPoints):
F = H + np.einsum('ikjl,k,l', Qt, C, C)
Eg = C.dot(H + F).dot(C) + 1. / X
energies[cycle] = Eg
if abs(oldE-Eg) < 1E-12:
break
Ct = (2. * mass * C - massMinusGamma * Cprev - 4. * F.dot(C) * dt2) / massPlusGamma
OC = O.dot(C)
OCt = O.dot(Ct)
OOC = O.dot(OC)
a = OOC.dot(OC) * dt4
b = -2. * OC.dot(OCt) * dt2
c = OCt.dot(Ct) - 1.
delta = b*b - 4.*a*c
if delta < 0:
print("Delta negative!")
break
sdelta = m.sqrt(delta)
lam1 = (-b-sdelta) / (2. * a)
lam2 = (-b+sdelta) / (2. * a)
if lam1 < 0:
lam = lam2
else:
lam = lam1
Ct -= lam * dt2 * OC
Cprev = C
C = Ct
oldE = Eg
It’s the code that relaxes the electrons towards the ground state. It’s used almost unchanged later in the code that also adds the dynamics of the nuclei, relaxing towards the equilibrium distance.
Conclusions
Please point out if you find any issues and have any suggestions. I’m also open to suggestions on what to add to the Python repository. I intend to add some more simple things in there sometime.
- PythonCompphys The python repository on GitHub ↩
- Car-Parrinello The notebook briefly described here ↩
- Computational Physics book by Jos Thijssen ↩
- Electronic Structure, Basic Theory and Practical Methods by Richard M Martin ↩
- Ab initio molecular dynamics lecture slides from University of Southampton ↩
- Unified Approach for Molecular Dynamics and Density-Functional Theory Phys. Rev. Lett. 55, 2471 – Published 25 November 1985, Car and Parrinello ↩
The post Car-Parrinello Quantum Molecular Dynamics first appeared on Computational Physics.

