Introduction
After some posts about the theory it is time to present the Hartree-Fock program. You might find the previous posts useful, along with the links in there: How to solve a quantum many-body problem, The Hartree-Fock method.
Here is the program in action (the final version might have minor differences):
Although it doesn’t show much, with minor changes you could do quite a bit more and with some effort a lot more. I had to stop development somewhere, though. I’m not happy with the code, especially about the IntegralsRepository, I feel that there is a lot to improve there, but here it is. At least it appears to work as intended and the code is pretty clear in the most important parts I wanted to present. This project took me more time than I anticipated, especially because of the electron-electron integrals. I think I had the restricted method working in less than a couple of hours (but it’s not the first time I implement a Hartree-Fock program), after having the integrals computation working, most of the difficulty was in having those integrals computed right. This includes finding clear enough papers on it, having the ability to test the results and other details you don’t take into account in advance.
Atoms and molecules
Obviously since the program deals with atoms and molecules, one should expect to have objects in the program that deal with them and indeed that is the case. Here is for example the class declaration for Atom:
class Atom
{
public:
Vector3D<double> position;
unsigned int Z;
unsigned int electronsNumber;
unsigned int ID;
Atom(unsigned int nrZ = 0, int nrElectrons = -1);
virtual ~Atom();
};
It contains what one would expect from it: a position for the atom, the atomic number, and the electrons number. The ID is there (and elsewhere) because it’s easier to identify a particular atom (or whatever has an ID) using an ID instead of its position, for example. The electrons number needs not to be equal with Z, it might be an ion, but it’s typically ignored and number of electrons for the whole molecule is used instead. There is an AtomWithShells derived from Atom which basically has shells assigned to it and some methods that deal with them.
Also a Molecule class exists, which as expected, has inside a bunch of atoms:
class Molecule
{
public:
std::vector<AtomWithShells> atoms;
unsigned int alphaElectrons;
unsigned int betaElectrons;
Molecule();
~Molecule();
unsigned int CountNumberOfContractedGaussians() const;
unsigned int CountNumberOfGaussians() const;
void SetIDs();
double NuclearRepulsionEnergy() const;
unsigned int ElectronsNumber();
unsigned int GetMaxAngularMomentum();
void SetCenterForShells();
void Normalize();
void Init();
};
All those classes are in the Systems namespace, to be grouped together and to be easier to locate.
The Basis
The program uses atomic orbitals as basis. More specifically, they are used to form a basis set which is not complete but it tries to span a subspace that includes most of the real solution.
Orbitals
The base class declaration looks like this:
class Orbital
{
public:
Vector3D<double> center;
QuantumNumbers::QuantumNumbers angularMomentum;
unsigned int ID;
unsigned int centerID;
unsigned int shellID;
Orbital();
virtual ~Orbital();
virtual double operator()(const Vector3D<double>& r) const = 0;
virtual Vector3D<double> getCenter() const;
char AtomicOrbital() const { return angularMomentum.AtomicOrbital(); }
};
It yet does not force you to use Gaussian-type orbitals (they could be Slater-type) or have them in Cartesian coordinates, they could be in spherical coordinates, for example (although the center vector holds Cartesian coordinates internally). The program uses Gaussian orbitals:
class GaussianOrbital : public Orbital
{
public:
double coefficient;
double alpha;
double normalizationFactor;
GaussianOrbital();
virtual ~GaussianOrbital();
virtual double getCoefficient() const;
virtual double getAlpha() const;
virtual double operator()(const Vector3D<double>& r) const;
Vector3D<double> ProductCenter(const GaussianOrbital& other) const;
protected:
double getNormalizationFactor() const;
public:
void Normalize();
};
By looking into GaussianOrbital::operator() you could find out that their value is:
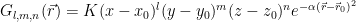
K is given by the normalization factor (the integral over the whole space should be 1) and a constant that is there because they are used in a linear combination to form a ‘contracted’ orbital that’s a better approximation to a Slater-type orbital. Here is the declaration for the contracted orbital:
class ContractedGaussianOrbital : public Orbital
{
public:
std::vector<GaussianOrbital> gaussianOrbitals;
virtual double operator()(const Vector3D<double>& r) const;
ContractedGaussianOrbital();
virtual ~ContractedGaussianOrbital();
void Normalize();
};
Several such orbitals are packed into a ‘contracted shell’:
class ContractedGaussianShell : public Shell
{
public:
std::vector<ContractedGaussianOrbital> basisFunctions;
ContractedGaussianShell();
~ContractedGaussianShell();
void AddOrbital(char type);
void AddGaussians(double exponent);
virtual Vector3D<double> getCenter() const;
std::string GetShellString() const;
unsigned int CountOrbitals(char orbital) const;
unsigned int CountContractedOrbitals(char orbital) const;
unsigned int CountNumberOfContractedGaussians() const;
unsigned int CountNumberOfGaussians() const;
virtual double operator()(const Vector3D<double>& r) const;
protected:
static unsigned int AdjustOrbitalsCount(char orbital, unsigned int res);
void AddOrbitalsInCanonicalOrder(unsigned int L);
public:
void SetCenters(const Vector3D<double>& center);
void Normalize();
};
The reason for packing them like that is to ease some integrals computations. An atom just has several such shells.
All orbitals are in the Orbitals namespace. There is also an important class in there, Orbitals::QuantumNumbers::QuantumNumbers. It packs the l, m, n integer values and it’s also a generator. That simplifies a lot iterating over integrals in the code, it’s more clear that way, but about that, later.
In the QuantumNumbers namespace there are also a lot of unused classes, like PxQuantumNumbers, PyQuantumNumbers and so on. I might use them later, they are there because I started to implement the code differently then I changed my mind, but still they could be useful in the future.
Basis
The basis is in the Chemistry namespace and it’s quite simple:
class Basis
{
public:
std::vector<Systems::AtomWithShells> atoms;
Basis();
~Basis();
void Load(std::string fileName);
void Save(const std::string& name);
void Normalize();
};
It just packs together a lot of atoms with their shells. They are loaded from a text file. The Load implementation is far from optimal and there is not much error checking, so it should be easy to crash with a malformed file. I just wanted to have something that works, I thought that I will polish the code afterwards but I gave up to the idea for now. It fulfills its purpose.
The source of the basis sets files is here: EMSL Basis Sets Exchange. Currently the program uses STO-3G and STO-6G basis sets, it could use other STO-nG with no change (and perhaps others, too). It should be able to use other Gaussian basis sets with no or minimal changes, too, but those should be enough for a program used as an example. If you want to generate another basis set to be used, be aware that I used NWChem format and I also replaced D+ and D- with E+ and E- in the file, to have the format that is easy to parse in C++ for double values. It should be very easy to change the program to read D+/- as well but I was lazy, ‘replace all’ appeared easier at that moment.
Matrices
As mentioned last time, matrix representation is used, the program deals with matrices a lot. The operators are matrices and that reflects in the implementation too. The Matrices namespace contains the matrices for overlap, kinetic and nuclear. They contain an Eigen matrix and have a Calculate method. They get the integrals from IntegralRepository (if not already calculated, they are calculated when retrieved). The implementation is easy, but you might have a little trouble to figure out what lines/columns represent. If you have two atoms with two shells each, that is, with 1s, 2s 2px 2py 2pz, then such a matrix will have 10 rows and 10 columns, the first 5 values being for the first atom, the next ones for the second one. For example the (0, 0) element of the S matrix will be 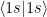 with 1s for the first atom for both bra and ket.
with 1s for the first atom for both bra and ket.
Integrals
Now for the most difficult part of the program… I will advise you to visit the links related to integrals from the previous post and I’ll add here more, specifically some that helped me with the implementation. First, the simple ones:
The last link is not so useful in the implementation part (the theory is!). You may recognize the recurrence relations in there, but the symbolic computation would give some troubles, also it uses numerical integration for Boys functions.
So here it is the main paper the implementation is based on, including the most complex integrals, the electron-electron ones: HSERILib: Gaussian integral evaluation. Fundamentals of Molecular Integrals Evaluation already given last time should also help. Another one that could help: Molecular Integrals over Gaussian Basis Functions.
The implementation is in GaussianIntegrals namespace. You’ll find there classes for each kind of integrals, overlap, kinetic, nuclear, electron-electron, along with some other classes such as the already mentioned IntegralsRepository which would really need an improvement.
If you look into the HSERILib paper for example and in the electron-electron integral code, you’ll notice that the recurrence formulae are as in the paper, with slight changes to fit computation better:
matrixCalc(curIndex, m) = RpaScalar * matrixCalc(prevIndex, m) + RwpScalar * matrixCalc(prevIndex, m + 1);
if (addPrevPrev)
{
unsigned int prevPrevIndex = prevPrevQN.GetTotalCanonicalIndex();
matrixCalc(curIndex, m) += N / (2. * alpha12) * (matrixCalc(prevPrevIndex, m) - alpha / alpha12 * matrixCalc(prevPrevIndex, m + 1));
}
Don’t forget that ‘quantum numbers’ are generators, the code might look simpler than it really is.
I won’t say anything more about the integrals, the code is on GitHub and the papers should be enough to understand it.
Hartree-Fock
The Hartree-Fock method is implemented in the HartreeFock namespace. Although I implemented the unrestricted method as well, I won’t present it here. The code is available, if interested you should check it out.
The restricted method is implemented into two classes, a base class, HartreeFockAlgorithm (also a base class for the unrestricted implementation) and the RestrictedHartreeFock class. This is how the self consistent field iteration is implemented:
double HartreeFockAlgorithm::Calculate()
{
double curEnergy = 0;
double prevEnergy = std::numeric_limits<double>::infinity();
if (!inited) return prevEnergy;
for (int iter = 0; iter < maxIterations; ++iter)
{
Step(iter);
curEnergy = GetTotalEnergy();
if (abs(prevEnergy - curEnergy) <= 1E-13) {
converged = true;
break;
}
if (terminate) break;
prevEnergy = curEnergy;
}
return curEnergy;
}
There is one other important method in there, HartreeFockAlgorithm::Init, but I’ll let you figure it out.
Here is the Step implementation for the restricted method:
void RestrictedHartreeFock::Step(int iter)
{
Eigen::MatrixXd F;
InitFockMatrix(iter, F);
Eigen::MatrixXd Fprime = Vt * F * V;
Eigen::SelfAdjointEigenSolver<Eigen::MatrixXd> es(Fprime);
const Eigen::MatrixXd& Cprime = es.eigenvectors();
Eigen::MatrixXd C = V * Cprime;
NormalizeC(C, nrOccupiedLevels);
Eigen::MatrixXd newP = Eigen::MatrixXd::Zero(h.rows(), h.cols());
for (int i = 0; i < h.rows(); ++i)
for (int j = 0; j < h.cols(); ++j)
for (unsigned int vec = 0; vec < nrOccupiedLevels; ++vec) newP(i, j) += 2. * C(i, vec) * C(j, vec);
const Eigen::VectorXd& eigenvals = es.eigenvalues();
CalculateEnergy(eigenvals, newP);
TRACE("Step: %d Energy: %f\n", iter, totalEnergy);
P = alpha * newP + (1. - alpha) * P; }
With the help of the comments and the theory from the last post, it should not be hard to understand. The NormalizeC call is not really necessary, the code should converge without it, but it might converge faster with normalization. In some implementations it is present, in others it is not. Physically, it makes sense to have the density matrix normalized. Probabilities should add up to 1. It should converge to such a density matrix anyway, but it makes sense to enforce a physical density matrix along the iterations, too.
The InitFockMatrix implementation is:
void RestrictedHartreeFock::InitFockMatrix(int iter, Eigen::MatrixXd& F) const
{
if (0 == iter)
{
if (initGuess > 0)
{
F.resize(h.rows(), h.cols());
for (int i = 0; i < h.rows(); ++i)
for (int j = 0; j < h.rows(); ++j)
F(i, j) = initGuess * overlapMatrix.matrix(i, j) * (h(i, i) + h(j, j)) / 2.;
}
else F = h;
}
else
{
Eigen::MatrixXd G = Eigen::MatrixXd::Zero(h.rows(), h.cols());
for (int i = 0; i < numberOfOrbitals; ++i)
for (int j = 0; j < numberOfOrbitals; ++j)
for (int k = 0; k < numberOfOrbitals; ++k)
for (int l = 0; l < numberOfOrbitals; ++l)
{
double coulomb = integralsRepository.getElectronElectron(i, j, k, l);
double exchange = integralsRepository.getElectronElectron(i, l, k, j);
G(i, j) += P(k, l) * (coulomb - 1. / 2. * exchange);
}
F = h + G;
}
}
For the 0 step, it simply sets the Fock matrix equal either with the ‘core’ matrix or with a ‘Hückel guess’. I won’t detail it more, but here is a start: Extended Hückel method.
The second part is more important, you can see how the Fock matrix is calculated out of the ‘core’ matrix (h = kinetic + nuclear) and density matrix P together with electron-electron interaction integrals (both coulomb and exchange terms). For the theory please visit the previous post.
That’s about it, the rest should be easy to figure out. The energy is calculated in RestrictedHartreeFock::CalculateEnergy.
The Program
Description
I usually list each class but now I have no patience for it, so I’ll describe it more briefly.
Classes
The most important classes that do not deal with user interface are in namespaces. The exceptions are HartreeFockThread derived from ComputationThread. Since the chart is composed from points which represent different computations, one can use several threads for computation. Anyway, even a single thread would make sense, to avoid locking the UI a long time. Vector3D is the same class seen in other projects on this blog, Chart is taken from another project that uses it on this blog, the other classes that are not in a namespace are quite similar with the ones in other projects, for details you could visit another post. The namespaces and classes in them were already briefly described above, except the Tensors namespace. I needed tensors for electron-electron calculation and although Eigen has some unsupported tensor implementation, I preferred to implement the classes myself. I might need them in a future project, too.
Libraries
As in other projects on this blog, besides mfc and other typical VC++ runtime libraries, the program uses GDI+ for drawing.
The program deals with matrices using Eigen library.
Classes usage
This is something that I quickly added to the repository readme file, I’ll also put it here, it might be useful:
Using the classes should be easy. Here is how to grab some atoms from the ‘basis’:
Systems::AtomWithShells H1, H2, O, N, C, He, Li, Ne, Ar;
for (auto &atom : basis.atoms)
{
if (atom.Z == 1) H1 = H2 = atom;
else if (atom.Z == 2) He = atom;
else if (atom.Z == 3) Li = atom;
else if (atom.Z == 8) O = atom;
else if (atom.Z == 6) C = atom;
else if (atom.Z == 7) N = atom;
else if (atom.Z == 10) Ne = atom;
else if (atom.Z == 18) Ar = atom;
}
Here is how to set the H2O molecule with the coordinates from the ‘Mathematica Journal’ (referenced in the code):
H1.position.X = H2.position.X = O.position.X = 0;
H1.position.Y = 1.43233673;
H1.position.Z = -0.96104039;
H2.position.Y = -1.43233673;
H2.position.Z = -0.96104039;
O.position.Y = 0;
O.position.Z = 0.24026010;
Systems::Molecule H2O;
H2O.atoms.push_back(H1);
H2O.atoms.push_back(H2);
H2O.atoms.push_back(O);
H2O.Init();
And here is how you calculate:
HartreeFock::RestrictedHartreeFock HartreeFockAlgorithm;
HartreeFockAlgorithm.alpha = 0.5;
HartreeFockAlgorithm.initGuess = 0;
HartreeFockAlgorithm.Init(&H2O);
double result = HartreeFockAlgorithm.Calculate();
You can do computation for a single atom, too, for now by putting it into a dummy molecule with a single atom in it. For example for He:
Systems::Molecule Heatom;
Heatom.atoms.push_back(He);
Heatom.Init();
Results
I tested the program with various molecules, the initial test was on the H2O molecule, but I also tested it with many more, comparing with Hartree-Fock limits and results from other programs.
I checked the intermediate results against the Mathematica Journal results, then I used other programs for tests (especially for electron-electron integrals). While doing that I found bugs in another program, too: bug 1 and bug 2, so I actually did more than implementing this project, I also helped identifying and fixing bugs in other project as well.
I’ll put here the chart in the featured image only, just as an example:
 CO2 molecule energy
CO2 molecule energy
Conclusion
This post ends the posts about Hartree-Fock. I might add a Post-Hartree-Fock method in the future, but certainly not this year. As usual, if you notice any mistakes/bugs please let me know.
The post The Hartree-Fock program first appeared on Computational Physics.

