Introduction
As promised in the Numerical Renormalization Group post, I implemented a Density Matrix Renormalization Group program. As I start writing this post, the program is already on GitHub. It’s quite basic, currently it is implemented only for Heisenberg model chains, for both spin 1/2 and 1. It runs only for even number of sites and symmetries are not used to speed up computation. Also it does not work for fermionic operators, although I might change it in the future to have it working for a Hubbard chain.
As for other posts, here is the program in action:
Currently it displays the ground state energy/site and the local bond strength. I checked if it is able to reproduce a result from Density-Matrix algorithms for quantum renormalization groups, fig 6a. One should be able to add ‘measurements’ for other operators fairly easy, either simple ones or long range correlations, this is only an example.
As usual, since I cannot cover the theory in a lot of details, I’ll give a lot of links that should help in understanding the program.
Links
On this blog
First, I’ll point to some related things that exist on this site. Since it’s a variational method, you might want to visit How to solve a quantum many body problem post. You might also want to visit the Renormalization Groups post for some generalities about renormalization. Very important is the The Numerical Renormalization Group. The idea for DMRG originated from there, and also DMRG ideas went back into NRG to make the Density Matrix Numerical Renormalization Group (which I briefly mentioned in that post, but you’ll find links in there with more details).
Lectures, PHD thesis
Here there are some lectures by Adrian Feiguin, with both theory and C++ code. Here is a dissertation: Competition of magnetic and superconducting ordering in one-dimensional generalized Hubbard models by Christian Dziurzik. There is an annex there that shows a way of handling fermionic operators signs. Here is the PhD thesis of Javier Rodriguez Laguna: Real Space Renormalization Group Techniques and Applications. A course by Andre Luiz Malvezzi: An introduction to numerical methods in low-dimensional quantum systems. And the last one in this paragraph: Density Matrix Renormalization Group for Dummies by Gabriele De Chiara, Matteo Rizzi, Davide Rossini, and Simone Montangero.
Reviews
A review paper by Ulrich Schollwoeck, The density-matrix renormalization group. A newer one by the same author The density-matrix renormalization group in the age of matrix product states, oriented as the title says, to Matrix Product States into which anybody who wants to implement more than a toy program must look.
Tutorials, codes
There are plenty of serious DMRG codes available, used for research. I’ll offer a link to only one of them: CheMPS2: a spin-adapted implementation of DMRG for ab initio quantum chemistry. You’ll find there not only the source code, but also links to publications, a workshop video and a user manual.
Since this blog is oriented to relatively small projects that are easier to understand than the sophisticated ones used for research, I’ll point to several simpler ones:
I found Tiny DMRG while looking for a Lanczos algoritm implementation. I had my implementation but because of the loss of orthogonality I looked for another one that has that prevented. I was simply too lazy to implement it myself, so I took the one from this project and changed it quite a bit to be easier to understand (also to use Eigen). Another pedagogical one is DMRG101, implemented in Python. This one is implemented in C++, requires armadillo: DMRGCat. This one implements Hubbard chains: Hubbard DMRG. I put the link here because for fermionic Hubbard chains one must take special care of fermionic operators (there is an annoying sign issue). I don’t know if I’m going to implement it in DMRG, if not, one can have a taste of it from the NRG project or look over this link to see how it’s done. Look also over the paper links above, it’s described in quite a detail. One more tutorial is Simple DMRG. One using Eigen and Matrix Product States is Eigen DMRG.
Another link that is worth a look is ITensor. You’ll find there – among others – info on Matrix Products Operators, Singular Value Decomposition and another way of dealing with fermionic operators, that is, Jordan-Wigner transformation.
Here is a lecture by Steven White:
Ok, those should suffice for now, I’m sure you can find more yourself if needed…
Density Matrix
Since the Density Matrix is an important concept for DMRG, I’ll try to write more about it here. I already mentioned it on the blog several times but I did not detail it much.
For a pure state
If the system is in a pure state 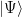 , then for an observable O we have a Hermitian operator
, then for an observable O we have a Hermitian operator  for which the expectation value is:
for which the expectation value is:
where the unit operator was inserted. is an operator and we note it with . It’s the Density Matrix operator for the pure state . We have:
You can immediately prove that the Density Matrix operator is a projection operator. Tr is the trace.
For a mixed state
Often instead of knowing a pure quantum state for a system, we have only a set of quantum states associated with probabilities for them. In that case, for an observable O we obtain the expected value as usual in the case with the classical probabilities:
is the probability of having the system in the pure state . is calculated as usual for the case of the pure quantum state . Density matrix turns into:
The Universe
To see how classical probabilities can arise from limited knowledge, let’s consider a system, called the Universe, composed of two systems, one called System, one Environment. Let’s say the system Hilbert space dimension is N and the environment Hilbert space dimension is M. The Hilbert space for the Universe has dimension NxM and it’s the tensor product of the two Hilbert spaces. Let’s assume that the Universe is in a pure quantum state, . The density matrix for it is . Writing
with
Considering the expectancy value of an observable that acts only on the System and does nothing to the Environment, that is:
after a bit of algebra one can find that:
With being the reduced density matrix obtained by tracing out the Environment:
The reduced density matrix describes a mixed state, if the System is entangled with the Environment.
Entanglement
The System is not entangled with the Environment if one can find that for and , , that is the pure state of the Universe can be written as a simple product between states of the System and Environment. If the Universe state cannot be expressed like that, the System and Environment are entangled.
How could you test if a density matrix describes a pure quantum state or a mixed one? For the above Universe case, how could one check if the reduced density matrix for the System describes an entangled state or a pure one? One way would be to notice that in the pure quantum state case . Another equivalent way is to find out is to find the rank (Schmidt rank, see below the details) of the matrix. If it’s 1, it’s a pure state. Another way to deal with it is Von Neumann entropy which is:
It’s easy to check that for a pure state the entropy is zero.
Schmidt decomposition
For an operator O represented by a nxn matrix, one can use eigendecomposition to switch to a basis where the operator is diagonal. For our description we have a ‘problem’, the System and the Environment do not necessarily have equal dimensions. There is a generalization of eigendecomposition which can be used, the Singular Value Decomposition. The generalization for eigenvalues is singular values. Applying the decomposition on the matrix of the coefficients of the pure function for the Universe, one obtains the Schmidt decomposition for the wave vector:
k runs up to . It means we can change the basis for both the System and the Environment to have the wave vector coefficients matrix diagonal. In this basis, called the Schmidt basis, tracing out the Environment gives the reduced density matrix for the System:
It’s similar when tracing out the System, the reduced density matrix of the Environment has the same eigenvalues as the one for the System, except that obviously the basis vectors are for the Environment instead of the System ones. Please check out the Lecture Notes of Adrian Feiguin for details.
The Problem
I will refer again the Numerical Renormalization Group post. I mentioned there that it works because of the exponential drop in the interaction strength. If one tries to apply the same method in real space to a chain, one can find out that it doesn’t work. You can’t simply add a site, diagonalize the Hamiltonian and pick up the states that have the lowest eigenvalues and drop the other ones, then repeat. Or better said, you can, but the results will not be very good. It turns out that the interaction with the environment/boundary conditions matter so much that they cannot be ignored while extending the chain. Wilson tried the blocks renormalization group on a single particle in a box and showed that it fails. For details please check the chapter 2 of Real Space Renormalization Group Techniques and Applications.
The Solution
Steven White published the solution in 1992: Density matrix formulation for quantum renormalization groups Steven R White, Phys. Rev. Lett. 69, 2863. The solution is based on two important things: do not ignore the environment and a better way of selecting the states that should be kept, than simply selecting those with the lowest energies.
The first one is obtained by considering not only the System as in NRG, grown one site each step, but also the Environment. The Environment can be obtained in the same way as the System, or if there is a mirror symmetry, one can simply reflect the System into the Environment. For the finite size algorithm, an Environment calculated at a previous step is used. This way not only the System is taken into account, but also the Environment and the interactions between the System and Environment, joined together into a superblock.
The selection of the best wave vectors when truncating the basis is done by considering the vectors that have a high probability. Considering the Schmidt decomposition and the expectancy value:
The operator is arbitrary, if we want an algorithm that keeps the most important states independent of it, we should keep the ones that correspond to the largest eigenvalues of the reduced density matrix, this way we should expect small errors.
Alternatively, one can see that if we want to obtain a good approximation for the whole pure wave vector , , we should try to minimize
For the coefficient matrices this is the Frobenius distance and the approximation is the low ranking approximation. For a given number of kept states r, the distance becomes:
Now it should be quite clear that we must retain the wave vectors corresponding to the largest eigenvalues of the reduced density matrix.
It turns out that this method also retains a maximum entanglement between System and Environment. Just calculate the von Neumann entropy and see how it changes by truncation.
That should be enough theory, from now on I’ll present things very briefly together with the code. For more details please check the links.
Infinite size
- The infinite size algorithm starts with a System block that could be formed from several sites, the code I implemented starts with a single site.
- Depending on what is implemented the System block can be reflected/copied into an Environment block (this is the case for the toy program, taking advantage of the mirror symmetry), or constructed in the same way as the System block, for more complex cases where one cannot take advantage of symmetry.
- Extend both the System and Environment blocks with a new site. The program I implemented just extends the System and then it copies it into the Environment block.
- Form a superblock from the System and Environment blocks, together with the interaction between them.
- Diagonalize the superblock Hamiltonian. Since we’re interested only in the ground state we can use a faster algorithm. The program uses the Lanczos algorithm with reorthonormalization. I implemented first myself the Lanczos one – I thought I could get away with it for this toy program – but it turned out that the orthogonality loss was big enough to need reorthogonalization, so I got another algorithm from GitHub and simplified it to be easier to understand for those that look over the code. You don’t need to use Lanczos, at first when implementing/testing the program I used a full diagonalization from Eigen, but it’s very slow compared with Lanczos. The Davidson or Arnoldi method could be also used, or any other method that takes advantage of the need for the ground state and ground energy only.
- Calculate the reduced density matrix.
- Diagonalize the reduced density matrix.
- Truncate the bases.
- Transform the Hamiltonian(s), the operators involved in the interaction and the operators for measurements in the new basis. This along with truncation is very similar with what is done in the Numerical Renormalization Group case, you might want to look over that one, too.
- Go again to extending the blocks and repeat the procedure until the ground energy does not change anymore appreciably or when you reach a certain size – when you continue with the finite size algorithm.
Here is how the infinite size algorithm looks in the code:
template<class SiteHamiltonianType, class BlockType> double GenericDMRGAlgorithm<SiteHamiltonianType, BlockType>::CalculateInfinite(int chainLength)
{
if (chainLength % 2) ++chainLength;
if (chainLength <= 0) return std::numeric_limits<double>::infinity();
ClearInit();
finiteAlgorithm = false;
double result = 0;
truncationError = 0;
for (int i = 0; 2 * systemBlock->length < chainLength; ++i)
result = Step(i);
return result;
}
Of course, the relatively complex work is in the Step call, but I’ll detail that later.
Finite size
- The finite size algorithm starts with the infinite size algorithm as described above, with the difference that both System and Environment blocks are saved, together with the ‘site’ operators in the blocks.
- If the desired size is reached, start performing sweeps.
- At first, the left block is increased as in the infinite size algorithm, but to preserve the superblock size, the Environment (right) block is decreased one site by using a previously saved smaller block.
- The site addition, superblock building, diagonalization, reduced density matrix calculation, truncating the basis and transforming operators is done as in the infinite size algorithm,
- Keep increasing the left block and decreasing the right one until the right one contains a single site, saving both left and right blocks each step.
- After reaching the right end, sweep towards the left. The toy program I implemented simply swaps the left and right and continues the algorithm as for the previously described left to right sweep.
- Repeat the sweeps until the ground state energy does not change appreciably anymore. Usually the sweep is ended in a symmetric configuration.
The algorithm can keep track of measurement operators, too. They have to be transformed, too. For details, please check out the documentation. The toy program implements it only for the left side blocks (for the last sweep), it can be implemented for both left and right but in this case I took advantage of the mirror symmetry.
Here is the finite size algorithm implementation:
template<class SiteHamiltonianType, class BlockType> double GenericDMRGAlgorithm<SiteHamiltonianType, BlockType>::CalculateFinite(int chainLength, int numSweeps)
{
if (chainLength % 2) ++chainLength;
if (chainLength <= 0) return std::numeric_limits<double>::infinity();
ClearInit();
finiteAlgorithm = false;
truncationError = 0;
systemBlocksRepository = new std::map<int, std::unique_ptr<BlockType>>();
environmentBlocksRepository = new std::map<int, std::unique_ptr<BlockType>>();
double result = 0;
AddToRepository(systemBlock->length, systemBlock, systemBlocksRepository);
AddToRepository(environmentBlock->length, environmentBlock, environmentBlocksRepository);
for (int i = 0; 2 * systemBlock->length < chainLength; ++i)
{
result = Step(i);
AddToRepository(systemBlock->length, systemBlock, systemBlocksRepository);
AddToRepository(environmentBlock->length, environmentBlock, environmentBlocksRepository);
}
bool left = false;
finiteAlgorithm = true;
for (int i = 0; i < numSweeps; ++i)
{
TRACE("SWEEP NUMBER %d\n", i);
for (int step = 0;; ++step)
{
int key = chainLength - systemBlock->length - 1;
delete environmentBlock;
environmentBlock = (*environmentBlocksRepository)[key].release();
environmentBlocksRepository->erase(key);
if (1 == environmentBlock->length)
{
std::swap(environmentBlock, systemBlock);
std::swap(environmentBlocksRepository, systemBlocksRepository);
left = !left;
AddToRepository(systemBlock->length, systemBlock, systemBlocksRepository);
if (!left && numSweeps - 1 == i) addInteractionOperator = true;
}
result = Step(step);
AddToRepository(systemBlock->length, systemBlock, systemBlocksRepository);
if (!left && systemBlock->length == chainLength / 2) break;
}
}
CalculateResults();
return result;
}
It’s a bit more complex but with the help of comments it should be relatively easy to understand. AddToRepository saves blocks, if (1 == environmentBlock->length) checks for swapping left with right.
The step detailed
Here is the code for Step:
template<class SiteHamiltonianType, class BlockType> double GenericDMRGAlgorithm<SiteHamiltonianType, BlockType>::Step(int step)
{
Extend();
if (!finiteAlgorithm) CopySystemBlockToEnvironment();
unsigned int SysBasisSize = (unsigned int)systemBlock->hamiltonian.matrix.cols();
unsigned int EnvBasisSize = (unsigned int)environmentBlock->hamiltonian.matrix.cols();
Operators::Hamiltonian superblockHamiltonian = CalculateSuperblock(SysBasisSize, EnvBasisSize);
double GroundStateEnergy = LanczosGroundState(superblockHamiltonian, GroundState);
TRACE(L"Energy/site: %f for system length: %d (Sys: %d, Env: %d) at step: %d\n", GroundStateEnergy / (systemBlock->length + environmentBlock->length), (systemBlock->length + environmentBlock->length), systemBlock->length, environmentBlock->length, step);
Operators::DensityMatrix densityMatrix(GroundState, SysBasisSize, EnvBasisSize);
densityMatrix.Diagonalize();
const Eigen::MatrixXd& eigenV = densityMatrix.eigenvectors();
eigenvals = densityMatrix.eigenvalues();
unsigned int keepStates = std::min<unsigned int>(maxStates, SysBasisSize);
Eigen::MatrixXd Ut(eigenV.rows(), keepStates);
int numStates = (int)eigenV.cols();
truncationError = 1.; for (unsigned int i = 0; i < keepStates; ++i)
{
int index = numStates - i - 1;
Ut.col(i) = eigenV.col(index);
truncationError -= eigenvals(index);
}
TRACE("Truncation Error: %f\n", truncationError);
const Eigen::MatrixXd U = Ut.adjoint();
TransformOperators(U, Ut);
return GroundStateEnergy;
}
With the help of the comments it should be easy to understand. Of course some of the work is delegated to other methods, either from the algorithm class or blocks. I’ll detail the most relevant ones later.
The Program
The program is quite simple and the mfc classes are not really interesting. It’s a standard mfc program so I won’t describe them anymore in detail. It’s just a doc/view program with options, property sheet and two property pages on it. Has a Chart which I took from another project already described on this blog. There is a ComputationThread that also resembles thread classes from other projects, from which DMRGThread is derived. The thread is started in CdmrgDoc::StartComputing() like this:
if (0 == theApp.options.model)
thread = new DMRGThread<DMRG::Heisenberg::DMRGHeisenbergSpinOneHalf>(theApp.options.sites, theApp.options.sweeps, theApp.options.states);
else
thread = new DMRGThread<DMRG::Heisenberg::DMRGHeisenbergSpinOne>(theApp.options.sites, theApp.options.sweeps, theApp.options.states);
thread->Start();
It would be quite easy to add another algorithm to it.
Please visit an older entry if you want to see a similar program described a little more. I’ll just describe here what’s different and related with DMRG.
Operators
Operators are implemented in the DMRG::Operators namespace. The root class is Operator but one is not supposed to use it directly. Two classes are derived from it, SiteOperator from which all ‘site’ operator classes are derived, and DiagonalizableOperator from which Hamiltonian and DensityMatrix operator classes are derived, obviously because we need diagonalization for them.
There is quite a bit of resemblance with the operator classes implemented in the Numerical Renormalization Group project, ideally they should share the classes but that’s not happening yet. There are differences, maybe in the future I’ll make them in such a way that they’ll be common. Since I mentioned NRG, I should mention that I kept a convention that is used there, that is, a newly added site (for the left block) is put in the most significant bits position in the Hamiltonian, that is, to the left. This way when the Hamiltonian is extended, the old Hamiltonian for the existing sites appears as blocks on the diagonal of the new matrix. I find it easier to visualize the newly added site (operators for it) in the matrix. It’s also easier to deal with the fermionic operators that way. Since currently the DMRG project implements only Heisenberg chains, it deals only with bosonic operators, so there is no sign issue. If you want to implement a Hubbard chain then you will have to deal with it. I already provided links that describe the issue in detail. You can also check out the Numerical Renormalization Group project and see how it’s handled there. Currently there is not much support in the DMRG project except that the Operator class has a changeSign flag that should indicate is the operator is fermionic, but there is no support in the implementation for it yet.
Please be aware that some tutorial projects and papers might extend the left Hamiltonian the other way around than in the convention I used, that is, I use for the left block while they might use .
Since I used tensor products in the formulae above, I should mention that the Kronecker products are implemented as static members in the Operator class. One implements the product between two matrices and the other two the product with the identity matrix.
There is not much else to say about the operators. I think the most important one to check is the DensityMatrix:
DensityMatrix::DensityMatrix(const Eigen::VectorXd& GroundState, unsigned int SysBasisSize, unsigned int EnvBasisSize, bool left)
: DiagonalizableOperator(SysBasisSize, false)
{
Eigen::MatrixXd c(EnvBasisSize, SysBasisSize);
for (unsigned int i = 0; i < EnvBasisSize; ++i)
for (unsigned int j = 0; j < SysBasisSize; ++j)
c(i, j) = GroundState(i * SysBasisSize + j);
if (left)
matrix = c.adjoint() * c;
else
matrix = c * c.adjoint();
}
Hopefully the comments are helpful. ‘Site’ operators are quite easy, for example for for 1/2 spin:
SzOneHalf::SzOneHalf(unsigned int size)
: SiteOperator(size, false)
{
int subsize = size / 2;
matrix.block(0, 0, subsize, subsize) = 1. / 2. * Eigen::MatrixXd::Identity(subsize, subsize);
matrix.block(subsize, subsize, subsize, subsize) = -1. / 2. * Eigen::MatrixXd::Identity(subsize, subsize);
}
It should be quite obvious that it is for:
Please check the project sources for the rest of operators implementation. There is not much more to it, really.
Blocks
I wrote a generic class for blocks, GenericBlock:
template<class SiteHamiltonianType> class GenericBlock
{
public:
GenericBlock(bool Left = true);
virtual ~GenericBlock();
int length;
bool left;
SiteHamiltonianType hamiltonian; SiteHamiltonianType SiteHamiltonian;
virtual void Extend();
virtual Operators::Hamiltonian GetInteractionHamiltonian() const = 0;
int GetSingleSiteBasisSize() const { return SiteHamiltonian.cols(); }
};
The GetInteractionHammiltonian is supposed to be implemented in a derived class. Extend should also be implemented in a derived class but the base class has an implementation for it:
template<class SiteHamiltonianType> void GenericBlock<SiteHamiltonianType>::Extend()
{
Operators::Hamiltonian interactionHamiltonian = GetInteractionHamiltonian();
hamiltonian.Extend(SiteHamiltonian, interactionHamiltonian, left);
++length;
}
That’s about it. For a specific model you just derive from it:
template<class SiteHamiltonianType, class SzType, class SplusType> class HeisenbergBlock : public GenericBlock<SiteHamiltonianType>
{
public:
HeisenbergBlock(bool Left = true);
virtual ~HeisenbergBlock();
SzType SzForBoundarySite;
SplusType SplusForBoundarySite;
static SzType SzForNewSite;
static SplusType SplusForNewSite;
virtual Operators::Hamiltonian GetInteractionHamiltonian() const;
virtual void Extend();
};
It contains operators needed for extending the block and implements the two methods and that’s about it.
Here is how it calculates the interaction Hamiltonian needed for adding a new site:
template<class SiteHamiltonianType, class SzType, class SplusType> Operators::Hamiltonian HeisenbergBlock<SiteHamiltonianType, SzType, SplusType>::GetInteractionHamiltonian() const
{
Operators::Hamiltonian interactionHamiltonian(hamiltonian.GetSingleSiteSize());
if (left)
{
interactionHamiltonian.matrix = Operators::Operator::KroneckerProduct(SzForNewSite.matrix, SzForBoundarySite.matrix) +
1. / 2. * (Operators::Operator::KroneckerProduct(SplusForNewSite.matrix, SplusForBoundarySite.matrix.adjoint()) +
Operators::Operator::KroneckerProduct(SplusForNewSite.matrix.adjoint(), SplusForBoundarySite.matrix));
}
else
{
interactionHamiltonian.matrix = Operators::Operator::KroneckerProduct(SzForBoundarySite.matrix, SzForNewSite.matrix) +
1. / 2. * (Operators::Operator::KroneckerProduct(SplusForBoundarySite.matrix, SplusForNewSite.matrix.adjoint()) +
Operators::Operator::KroneckerProduct(SplusForBoundarySite.matrix.adjoint(), SplusForNewSite.matrix));
}
return interactionHamiltonian;
}
and here is how the Extension is extended (sic):
template<class SiteHamiltonianType, class SzType, class SplusType> void HeisenbergBlock<SiteHamiltonianType, SzType, SplusType>::Extend()
{
int BasisSize = (int)hamiltonian.matrix.cols();
GenericBlock::Extend();
if (left)
{
SplusForBoundarySite.matrix = Operators::Operator::KroneckerProductWithIdentity(SplusForNewSite.matrix, BasisSize);
SzForBoundarySite.matrix = Operators::Operator::KroneckerProductWithIdentity(SzForNewSite.matrix, BasisSize);
}
else
{
SplusForBoundarySite.matrix = Operators::Operator::IdentityKronecker(BasisSize, SplusForNewSite.matrix);
SzForBoundarySite.matrix = Operators::Operator::IdentityKronecker(BasisSize, SzForNewSite.matrix);
}
}
This concludes the blocks discussion. I should mention two things, though.
First, it’s usually safe to ignore code like if (left) in this project (there might be a couple of places where it’s not) and consider only the code in the if (left) branch ignoring the else part. The reason is that the right block is simply copied from the left one. I added the ‘left’ check just in case the code will be extended to construct the right block in a similar manner as the left one.
Second, you might be a little confused by the block copy. It should be reflected, right? You may visualize what’s happening by considering the two left and right blocks with the two sites in between (as in the featured image but symmetrical). Bend the chain in the middle until the right block is turned under the left one. Together with a relabeling of the sites in the right (now under) block, it should be more clear what is happening.
Algorithm implementation
I already presented the most important parts of the algorithm implementation above, but there is more to it. I implemented a GenericDMRGAlgorithm where most of the code is, the model specific code being the responsibility of derived classes. This class is the most important but I cannot present it here entirely. Please check the source code for details about it. What I presented here about it should be enough to help you understand it. Just one more thing about it here:
template<class SiteHamiltonianType, class BlockType> Operators::Hamiltonian GenericDMRGAlgorithm<SiteHamiltonianType, BlockType>::CalculateSuperblock(unsigned int SysBasisSize, unsigned int EnvBasisSize)
{
Operators::Hamiltonian superblockHamiltonian;
Operators::Hamiltonian SystemEnvironmentInteraction = GetInteractionHamiltonian();
superblockHamiltonian.matrix = Operators::Operator::IdentityKronecker(EnvBasisSize, systemBlock->hamiltonian.matrix) +
SystemEnvironmentInteraction.matrix +
Operators::Operator::KroneckerProductWithIdentity(environmentBlock->hamiltonian.matrix, SysBasisSize);
return superblockHamiltonian;
}
That’s how the Superblock Hamiltonian is obtained.
From this generic class I derived the Heisenberg class. It does not have much into it, it just implements a couple of methods. One is for the interaction Hamiltonian between the two blocks (needed when constructing the Superblock):
template<class SiteHamiltonianType, class SzType, class SplusType> Operators::Hamiltonian HeisenbergDMRGAlgorithm<SiteHamiltonianType, SzType, SplusType>::GetInteractionHamiltonian() const
{
Operators::Hamiltonian interactionHamiltonian;
interactionHamiltonian.matrix = Operators::Operator::KroneckerProduct(environmentBlock->SzForBoundarySite.matrix, systemBlock->SzForBoundarySite.matrix) +
1. / 2. * (Operators::Operator::KroneckerProduct(environmentBlock->SplusForBoundarySite.matrix, systemBlock->SplusForBoundarySite.matrix.adjoint()) +
Operators::Operator::KroneckerProduct(environmentBlock->SplusForBoundarySite.matrix.adjoint(), systemBlock->SplusForBoundarySite.matrix));
return interactionHamiltonian;
}
The other one is for transforming the operators in the new basis:
template<class SiteHamiltonianType, class SzType, class SplusType> void HeisenbergDMRGAlgorithm<SiteHamiltonianType, SzType, SplusType>::TransformOperators(const Eigen::MatrixXd& U, const Eigen::MatrixXd& Ut, bool left)
{
GenericDMRGAlgorithm::TransformOperators(U, Ut, left);
if (left)
{
systemBlock->SplusForBoundarySite.matrix = U * systemBlock->SplusForBoundarySite.matrix * Ut;
systemBlock->SzForBoundarySite.matrix = U * systemBlock->SzForBoundarySite.matrix * Ut;
}
else
{
environmentBlock->SplusForBoundarySite.matrix = U * environmentBlock->SplusForBoundarySite.matrix * Ut;
environmentBlock->SzForBoundarySite.matrix = U * environmentBlock->SzForBoundarySite.matrix * Ut;
}
}
Not a big deal, the call into the base class does something similar for the Hamiltonian and measurement operators, that is .
And this is the final class for Heisenberg spin 1/2:
class DMRGHeisenbergSpinOneHalf :
public HeisenbergDMRGAlgorithm<Operators::Hamiltonian, Operators::SzOneHalf, Operators::SplusOneHalf>
{
public:
DMRGHeisenbergSpinOneHalf(unsigned int maxstates = 18);
};
There isn’t much to it, the ‘trick’ is in the template arguments and that’s about it. Here is the constructor:
DMRGHeisenbergSpinOneHalf::DMRGHeisenbergSpinOneHalf(unsigned int maxstates)
: HeisenbergDMRGAlgorithm<Operators::Hamiltonian, Operators::SzOneHalf, Operators::SplusOneHalf>(maxstates)
{
}
That’s the end of the algorithm presentation. There isn’t much more in the code, except in GenericDMRGAlgorithm. The most important method that I did not present here is LanczosGroundState. With the help of the comments it should be easy to understand. You might want to also check Extend, TransformOperators and CalculateResults. They are only a few lines long and easy to figure out.
When you are looking over the GitHub code you might find out that it’s not identical with what I present here. It might be the case that I improved/extended the code, but this post will more or less stay as it is. I have to warn you about possible differences, I might change the code in the future (also the NRG one).
Libraries
As for other projects before, this project uses mfc, the chart draws using GDI+ and the matrix library used is Eigen.
Results
I used Density-Matrix algorithms for quantum renormalization groups, by Steven R White to check if the program works correctly. It seems that the program reproduces correctly fig 6a (spin 1/2, 60 sites) and fig 7a (spin 1, 60 sites). With some minimal changes it should be able to reproduce all charts from fig 6 and 7.
Here is the one for fig 6, only half of it because of the mirror symmetry:
Spin 1/2, 60 sites
Possible Improvements
I’ll indicate here some possible improvements, they won’t be exhaustive. The subject is very large, please check the links I provided for more info. If you intend to write a serious program, you should look into matrix product states.
There are some easy things that can be done even for this toy program and some not so easy.
- Mirror symmetry allows you to avoid a full sweep, half sweep is enough. The program could be changed to do that (or not) depending on a ‘mirror symmetry’ flag. It’s not a big deal.
- Change the code to build the ‘right’ block too, if there is no mirror symmetry. This should be very easy, part of the code is already in place.
- The code currently deals with measurement operators only in the left block and only the interaction ones. This could be made more general and could also allow operators for the right block, or two-point correlators that act on both blocks, too.
- Change the code to allow odd number of sites. Although it’s very tempting to just put a site in the middle when constructing the superblock, don’t do it! The Hamiltonian matrix gets too large and it slows the program. What can be done is to use an Environment block that is a site less than the System block. That even allows you to reuse the System block from the previous step as the Environment, during the infinite size algorithm.
- This should be also easy: Currently the Heisenberg model is with J=1. One could provide two couplings, one for the Ising part one for the ‘xy’ part of the interaction, this allows computation for various couplings, including for example both antiferromagnetic and ferromagnetic Ising model.
- Currently the code considers only the ground state and energy. You could extend the code to consider several lowest levels. If you are interested only in the gap between the ground state and the first excited state, you could use the algorithm almost unchanged, executed twice, once as usual and once for a modified Hamiltonian , where is the previously obtained ground state. This pushes up the old ground state (with an energy given by k) so the first excitation state (this discussion assumes the ground state not being degenerate, which is not always the case) becomes the new ground state. Another way is to target several states during a single algorithm run, and use a density matrix which is an average of density matrices for the individual states (as in where for example for two states w could be simply 1/2).
If you implement symmetries, your model can have the first (interesting) excited state in a different sector than the one that contains the ground state, this eases things up. This can be a good way of calculating a band gap. - Allow inserting an ‘impurity’ in the chain.
- Improve the Lanczos algorithm. Perhaps with sparse matrices? Currently it’s an easy implementation that does not perform as well as it could.
- Wavefunction prediction: Currently during a sweep the Lanczos tridiagonalization starts from a random wave vector. The performance can be improved by using the wave vector from the latest step to make a guess for the initial wave vector for the Lanczos algorithm. More info here.
- tDMRG. Lecture Notes by Adrian Feiguin describes an implementation.
- Add a Hubbard chain implementation. Since it involves fermionic operators, this has a sign problem that has to be taken into account. A good start is the implementation of NRG – it has to deal with fermionic operators – I have for this blog and there is also information in the links I provided.
- Consider models that have more than nearest site interaction. This makes the site additions and constructing the superblock a bit more complex.
- Consider not only chains but also ladders.
- Momentum space model?
- Are you brave enough for Quantum Chemistry or even Nuclear Physics? In Quantum Chemistry the sites are orbitals (as in Hartree-Fock or Density Functional Theory, please check out the Hartree-Fock posts for details) and in Nuclear Physics the sites are the nuclear shells.
- Last but not least (this is actually easier than some of the above) you should consider symmetries to speed up calculations. I mentioned this in the Numerical Renormalization Group post, I’ll briefly mention here again: with symmetries you end up with the Hamiltonian being block diagonal. Instead of dealing with the large matrix you deal with the small ones. Operators in general couple different quantum numbers and they are not block diagonal, but you still can deal with the small blocks instead of the whole matrix. This is an important issue, perhaps I should change either NRG or DMRG project to have this exemplified (perhaps both?).
There is much more to investigate, please at least check out the The density-matrix renormalization group review by Ulrich Schollwoeck for more that could be done and more details about the above hints.
Conclusion
That’s the end for DMRG for now. I might have some additions in the future for it, though.
As usual, if you notice any mistakes/bugs or you have suggestions, please let me know.
Later changes to the code
I won’t update the code description above, I’ll try to keep it simple, but the code can suffer changes over time.
Currently the code has the following additions:
- Couplings for the Heisenberg model can be specified
- It can deal with odd number of sites.
- It can handle more states, not only the ground state. I didn’t test it much, but it seems to be able to obtain the Haldane energy gap for the Heisenberg chain with spin 1. I’ll have to test it with a longer chain and more eigenvectors retained to be sure. I picked the easiest solution for implementing this, the code is delimited in the
Step implementation, so the added complexity should not make the code harder to understand.
The post Density Matrix Renormalization Group first appeared on Computational Physics.

